Pharmacotherapeutic group: Drugs for obstructive airway diseases, anticholinergics.
ATC code: R03BB07.
Pharmacology: Pharmacodynamics: Mechanism of action: Umeclidinium bromide is a long acting muscarinic receptor antagonist (also referred to as an anticholinergic). It is a quinuclidine derivative that is a muscarinic receptor antagonist with activity across multiple muscarinic cholinergic receptor subtypes. Umeclidinium bromide exerts its bronchodilatory activity by competitively inhibiting the binding of acetylcholine with muscarinic cholinergic receptors on airway smooth muscle. It demonstrates slow reversibility at the human M3 muscarinic receptor subtype
in vitro and a long duration of action
in vivo when administered directly to the lungs in pre-clinical models.
Pharmacodynamic effects: In a Phase III, 6-month study (DB2113373) INCRUSE provided a clinically meaningful improvement over placebo in lung function (as measured by forced expiratory volume in 1 second [FEV
1]) over 24 hours following once daily administration, which was evident at 30 minutes following administration of the first dose (improvement over placebo by 102 mL, p<0.001*). The mean peak improvements in FEV
1 within the first 6 hours following dosing relative to placebo were 130 ml (p<0.001*) at Week 24. There was no evidence for tachyphylaxis in the effect of INCRUSE over time.
Cardiac electrophysiology: The effect of umeclidinium 500 micrograms (pre-dispensed) on the QT interval was evaluated in a placebo- and moxifloxacin-controlled QT trial of 103 healthy volunteers. Following repeat doses of umeclidinium 500 micrograms once daily for 10 days, no clinically relevant effect on prolongation of QT interval (corrected using the Fridericia method) or effects on heart rate were observed.
Clinical efficacy: The clinical efficacy of INCRUSE administered once daily was evaluated in 904 adult patients who received umeclidinium bromide or placebo from two pivotal Phase III clinical studies with a clinical diagnosis of COPD; a 12-week study (AC4115408) and a 24-week study (DB2113373).
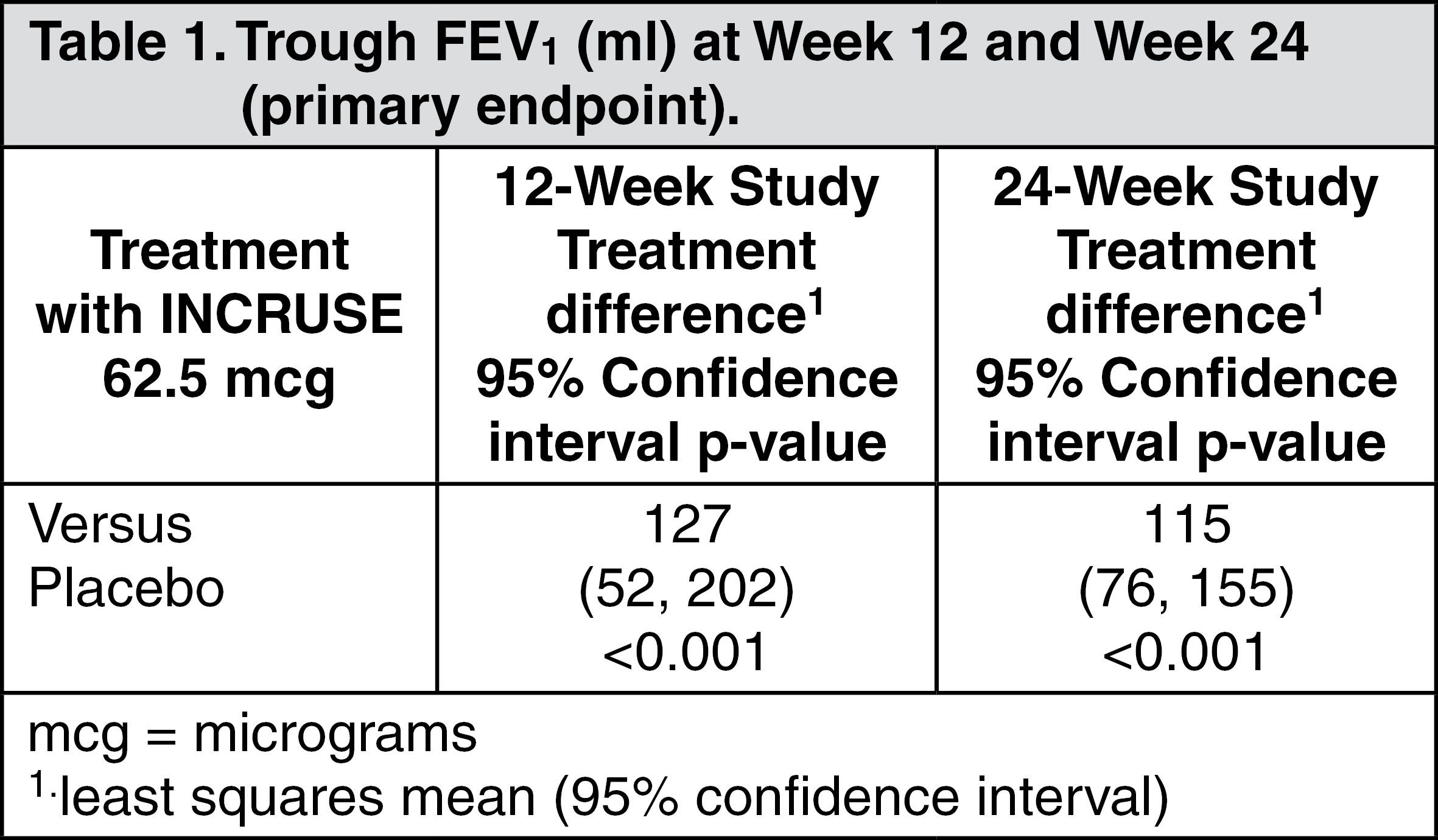
Pivotal Efficacy Studies: Effects on lung function: In both of the pivotal 12-week and 24-week studies, INCRUSE demonstrated statistically significant and clinically meaningful improvements in lung function (as defined by change from baseline trough FEV
1 at Week 12 and Week 24 respectively, which was the primary efficacy endpoint in each study) compared with placebo (see Table 1). The bronchodilatory effects with INCRUSE compared with placebo were evident after the first day of treatment in both studies and were maintained over the 12-week and 24-week treatment periods.
There was no attenuation of the bronchodilator effect over time. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
INCRUSE demonstrated a statistically significant greater improvement from baseline in weighted mean FEV
1 over 0-6 hours post-dose at Week 12 compared with placebo (166 ml, p<0.001) in the 12-week pivotal study. INCRUSE demonstrated a greater improvement from baseline in weighted mean FEV
1 over 0-6 hours post-dose at Week 24 compared with placebo (150 ml, p<0.001*) in the 24-week pivotal study.
Symptomatic outcomes: Breathlessness: In the 12-week study, a statistically significant improvement compared with placebo in the TDI focal score at Week 12 was not demonstrated for INCRUSE (1.0 units, p=0.05).
A statistically significant improvement compared with placebo in the TDI focal score at Week 24 was demonstrated for INCRUSE (1.0 units, p<0.001) in the 24-week study.
The proportion of patients who responded with at least the minimum clinically important difference (MCID) of 1 unit TDI focal score at Week 12 was greater for INCRUSE (38%) compared with placebo (15%) in the 12-week study. Similarly, a greater proportion of patients achieved ≥1 unit TDI focal score for INCRUSE (53%) compared with placebo (41%) at Week 24 in the 24-week study.
Health-related quality of life: INCRUSE also demonstrated a statistically significant improvement in health-related quality of life measured using the St. George's Respiratory Questionnaire (SGRQ) as indicated by a reduction in SGRQ total score at Week 12 compared with placebo (-7.90 units, p<0.001) in the 12-week study. A greater improvement compared with placebo in the change from baseline in SGRQ total score at Week 24 was demonstrated for INCRUSE (-4.69 units, p<0.001*) in the 24-week study.
The proportion of patients who responded with at least the MCID in SGRQ score (defined as a decrease of 4 units from baseline) at Week 12 was greater for INCRUSE 62.5 micrograms (44%) compared with placebo (26%) in the 12-week study. Similarly, a greater proportion of patients achieved at least the MCID for INCRUSE at Week 24 (44%) compared with placebo (34%) in the 24-week study.
COPD exacerbations: In the 24-week study, INCRUSE lowered the risk of a COPD exacerbation compared with placebo (analysis of time to first exacerbation; Hazard Ratio 0.6, p=0.035*). The probability of having an exacerbation in patients receiving INCRUSE at week 24 was 8.9% compared with 13.7% for placebo. These studies were not specifically designed to evaluate the effect of treatments on COPD exacerbations and patients were withdrawn from the study if an exacerbation occurred.
Use of rescue medicinal product: In the 12-week study, INCRUSE statistically significantly reduced the use of rescue medication with salbutamol compared with placebo (on average a reduction of 0.7 puffs per day over Weeks 1-12, p=0.025) and demonstrated a higher percentage of days when no rescue medication was needed (on average 46.3%) compared with placebo (on average 35.2%; no formal statistical analysis was performed on this endpoint). In the 24-week study treatment with INCRUSE, the mean (SD) change from baseline in the number of puffs of rescue salbutamol over the 24-week treatment period was -1.4 (0.20) for placebo and -1.7 (0.16) for INCRUSE (Difference = -0.3; 95% CI: -0.8, 0.2, p=0.276). Patients receiving INCRUSE had a higher percentage of days when no rescue medication was needed (on average 31.1%) compared with placebo (on average 21.7%). No formal statistical testing was performed on this endpoint.
Supporting efficacy studies: In two 12-week, placebo controlled studies (200109 and 200110), the addition of Incruse to fluticasone furoate/vilanterol (FF/VI) (Relvar Ellipta 100/25 micrograms) once daily in adult patients with a clinical diagnosis of COPD, resulted in statistically significant and clinically meaningful improvements in the primary endpoint of trough FEV
1 at Day 85 compared to placebo plus FF/VI (124 mL (95% CI 93, 154, p<0.001) and 122 mL (95%CI 91, 152, p<0.001)).
Improvements in lung function were supported with reductions in use of salbutamol over Weeks 1-12 (-0.4 puffs per day (95% CI -0.7, -0.2, p<0.001) and -0.3 puffs per day (95% CI -0.5, -0.1, p=0.003)) compared to placebo plus FF/VI but improvements in SGRQ at week 12 were not statistically significant (200109) or clinically relevant (200109 and 200110). The short duration of the studies and limited number of exacerbation events, preclude any conclusion regarding additional effect of Incruse on COPD exacerbation rate.
No new adverse drug reactions were identified with the addition of Incruse to FF/VI in these studies.
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with INCRUSE in all subsets of the paediatric population in COPD (see Dosage & Administration for information on paediatric use).
*A step-down statistical testing procedure was used in this study and this comparison was below a comparison that did not achieve statistical significance. Therefore, statistical significance on this comparison cannot be inferred.
Pharmacokinetics: Absorption: Following inhaled administration of umeclidinium bromide in healthy volunteers, C
max occurred at 5 to 15 minutes. The absolute bioavailability of inhaled umeclidinium bromide was on average 13% of the dose, with negligible contribution from oral absorption. Following repeat dosing of inhaled umeclidinium bromide, steady state was achieved within 7 to 10 days with 1.5 to 1.8-fold accumulation.
Distribution: Following intravenous administration to healthy subjects, the mean volume of distribution was 86 litres.
In vitro plasma protein binding in human plasma was on average 89%.
Biotransformation: In vitro studies showed that umeclidinium bromide is principally metabolised by cytochrome P450 2D6 (CYP2D6) and is a substrate for the P-glycoprotein (P-gp) transporter. The primary metabolic routes for umeclidinium bromide are oxidative (hydroxylation, O-dealkylation) followed by conjugation (glucuronidation, etc), resulting in a range of metabolites with either reduced pharmacological activity or for which the pharmacological activity has not been established. Systemic exposure to the metabolites is low.
Elimination: Plasma clearance following intravenous administration was 151 litres/hour. Following intravenous administration, approximately 58% of the administered radiolabelled dose (or 73% of the recovered radioactivity) was excreted in faeces by 192 hours post-dose. Urinary elimination accounted for 22% of the administered radiolabelled dose by 168 hours (27% of recovered radioactivity). The excretion of the drug-related material in the faeces following intravenous dosing indicated secretion into the bile. Following oral administration to healthy male subjects, total radioactivity was excreted primarily in faeces (92% of the administered radiolabelled dose or 99% of the recovered radioactivity) by 168 hours post-dose. Less than 1% of the orally administered dose (1% of recovered radioactivity) was excreted in urine, suggesting negligible absorption following oral administration. Umeclidinium bromide plasma elimination half-life following inhaled dosing for 10 days averaged 19 hours, with 3% to 4% active substance excreted unchanged in urine at steady-state.
Characteristics in specific groups of subjects or patients: Elderly: A population pharmacokinetic analysis showed that pharmacokinetics of umeclidinium bromide are similar between COPD patients 65 years and older and those younger than 65 years of age.
Renal impairment: Subjects with severe renal impairment (creatinine clearance <30mL/min) showed no evidence of an increase in systemic exposure to umeclidinium bromide (C
max and AUC), and no evidence of altered protein binding between subjects with severe renal impairment and healthy volunteers.
Hepatic impairment: Subjects with moderate hepatic impairment (Child-Pugh Class B) showed no evidence of an increase in systemic exposure to umeclidinium bromide (C
max and AUC), and no evidence of altered protein binding between subjects with moderate hepatic impairment and healthy volunteers. Umeclidinium bromide has not been evaluated in subjects with severe hepatic impairment.
Other special populations: A population pharmacokinetic analysis showed that no dose adjustment is required for umeclidinium bromide based on the effect of age, race, gender, inhaled corticosteroid use or weight. A study in CYP2D6 poor metabolisers showed no evidence of a clinically significant effect of CYP2D6 genetic polymorphism on systemic exposure to umeclidinium bromide.
Toxicology: Preclinical safety data: Non clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and carcinogenic potential. In nonclinical studies with umeclidinium bromide, findings were those typically associated with the primary pharmacology of muscarinic receptor antagonists and/or local irritancy.
Reproductive toxicity: Umeclidinium bromide was not teratogenic in rats or rabbits. In a pre- and post-natal study, subcutaneous administration of umeclidinium bromide to rats resulted in lower maternal body weight gain and food consumption and slightly decreased pre-weaning pup body weights in dams given 180 micrograms/kg/day dose (approximately 80-times the human clinical exposure of umeclidinium 62.5 micrograms, based on AUC).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 62.5 mcge59d85b8-5f2c-4aa1-bc58-a8dd010a4a95.GIF)

 Sign Out
Sign Out
